Data preprocessing
In this tutorial, we demonstrate how to prepare the required inputs for training the SVC model, using seqFISH+ data from Eng et al. (2019) as an example.
Preprocessed datasets used in our project that are ready to use can be downloaded from: https://drive.google.com/drive/folders/1G4Nl-As6hXsAh7vHaRJ3vkYv44A-mU5f
import warnings
warnings.filterwarnings("ignore")
from tqdm import tqdm
import scanpy as sc
from model.utils import *
import natsort
import matplotlib.pyplot as plt
load subcellular ST data
x y: Cartesian coordinates of a measured transcript within the cellgene: Gene name of the transcriptcell: Cell ID that the transcript belongs tocenterX centerY: Nuclear center coordinates for the corresponding cell
data_path = './SVC/'
dataset = 'data/seqfish'
data = pd.read_pickle(f"{data_path}{dataset}/seqfish_data_dict.pkl")
seqfish_data = data['data_df']
print(seqfish_data.head())
x y gene cell nucleus batch umi centerX \
0 1217.437557 557.583252 4933401b06rik 5-0 -1 0 1 1003
1 1096.190309 394.835294 4933401b06rik 5-0 5 0 1 1003
2 1093.189494 572.832405 4933401b06rik 5-0 -1 0 1 1003
3 1005.120220 297.196271 4933401b06rik 5-0 -1 0 1 1003
4 1142.815026 378.376491 4933401b06rik 5-0 -1 0 1 1003
centerY type sc_total
0 425 fibroblast 32224
1 425 fibroblast 32224
2 425 fibroblast 32224
3 425 fibroblast 32224
4 425 fibroblast 32224
Gene2vec ortholog mapping and gene filtering
For mouse data, we mapped the 16,906 Gene2vec genes to mouse orthologs and preserve the common genes.
ref_gene_names = np.loadtxt(f"{data_path}/data/gene2vec/convert_mmu_gene2vec_names.txt", dtype=str).tolist() ## for human data, use gene2vec/gene2vec_names.txt instead
common= list(set(np.array(list(data['gene_list_dict'].values()))[0]).intersection(set(ref_gene_names)))
print("common genes:", len(common))
common genes: 2659
We then selected the top 1,000 most highly expressed genes for model training and downstream analyses.
seqfish_data = seqfish_data[seqfish_data.gene.isin(common)]
high_exp_gene = seqfish_data.groupby(['gene']).count()['x'].sort_values(ascending=False)[:1000]
high_exp_gene_id = np.array(high_exp_gene.index)
gene_names = natsort.natsorted(high_exp_gene_id)
seqfish_data = seqfish_data[seqfish_data.gene.isin(gene_names)]
print("cell number:",seqfish_data.cell.unique().shape[0])
print("gene number:",seqfish_data.gene.unique().shape[0])
# np.savetxt(f"{data_path}{dataset}/gene_names.txt", gene_names, fmt='%s')
cell_names = natsort.natsorted(seqfish_data['cell'].unique().tolist())
# np.savetxt(f"{data_path}{dataset}/cell_names.txt", cell_names, fmt='%s')
cell number: 171
gene number: 1000
save the corresponding Gene2Vec vectors
chosen_indices_ref = []
for i in range(len(gene_names)):
if gene_names[i] in ref_gene_names:
index_position = ref_gene_names.index(gene_names[i])
chosen_indices_ref.append(index_position)
gene2vec_weight = np.load(f'{data_path}/data/gene2vec/gene2vec_16906.npy')
print("gene2vec_weight:", gene2vec_weight.shape)
gene2vec_weight_filtered = gene2vec_weight[chosen_indices_ref]
print("filtered gene2vec_weight:", gene2vec_weight_filtered.shape)
# np.save(f"{data_path}{dataset}/gene2vec_weight_seqfish.npy", gene2vec_weight_filtered)
gene2vec_weight: (16906, 200)
filtered gene2vec_weight: (1000, 200)
load cell boundary information for each cell
direction_vec: the angle of transcript relative to the positive x-axis, with the nuclear center as the origin.distance_to_center: Euclidean distance between the cell boundary point and nuclear center
cell_mask_contour = pd.read_pickle(f"{data_path}{dataset}/cell_mask_contour_preprocessed.pkl")
print(cell_mask_contour.head())
cell x y centerX centerY direction_vec distance_to_center
0 0-0 521 496 1079 724 -158.0 602.783543
1 0-0 519 496 1079 724 -158.0 604.635427
2 0-0 517 494 1079 724 -157.5 607.242950
3 0-0 515 494 1079 724 -158.0 609.094410
4 0-0 513 492 1079 724 -157.5 611.702542
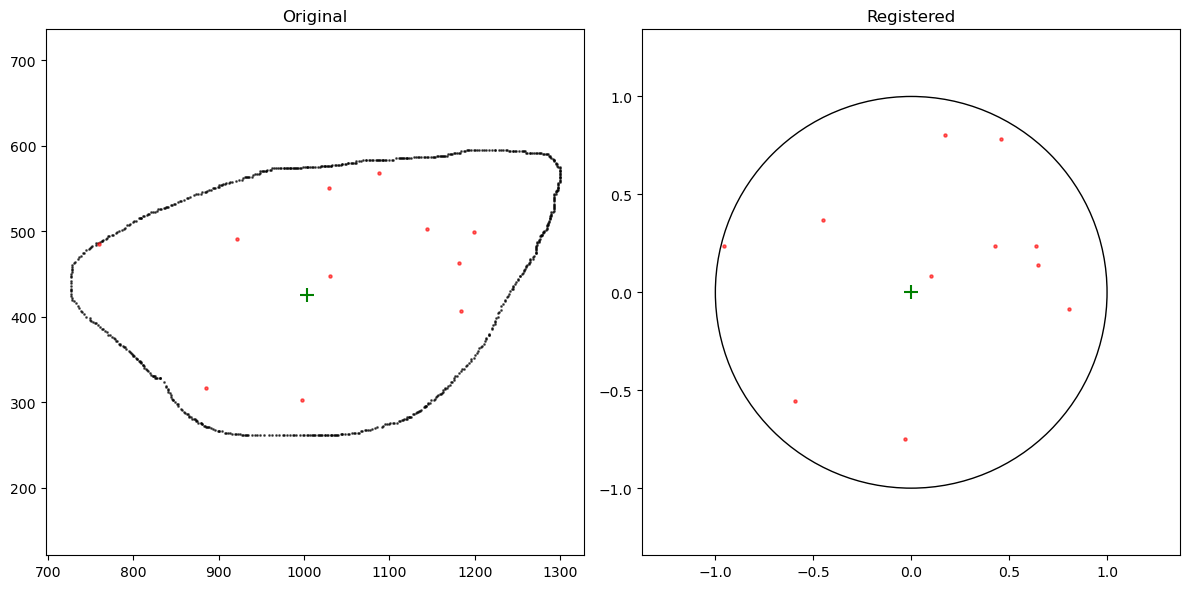
Conduct cell registration procedure
seqfish_data = register_original_data(seqfish_data, cell_mask_contour)
100%|██████████| 171/171 [07:26<00:00, 2.61s/it]
x y gene cell nucleus batch umi centerX centerY \
10 828.109167 457.736354 Aatf 5-0 -1 0 1 1003 425
11 1128.101035 488.990636 Aatf 5-0 -1 0 1 1003 425
12 1039.856556 294.494083 Aatf 5-0 -1 0 1 1003 425
13 1182.850103 461.492268 Aatf 5-0 -1 0 1 1003 425
14 891.607843 440.987851 Aatf 5-0 5 0 1 1003 425
type sc_total distance_to_center direction_vec ratio \
10 fibroblast 32224 177.928278 169.5 0.667541
11 fibroblast 32224 140.517153 27.0 0.425739
12 fibroblast 32224 135.610472 -74.0 0.802344
13 fibroblast 32224 183.514973 11.5 0.668383
14 fibroblast 32224 112.533657 172.0 0.409745
angle_radians x_norm y_norm
10 2.958333 -0.656363 0.121650
11 0.471239 0.379336 0.193281
12 -1.291544 0.221156 -0.771263
13 0.200713 0.654965 0.133254
14 3.001966 -0.405758 0.057026
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
cell_mask_contour_i = cell_mask_contour[(cell_mask_contour.cell == '5-0')]
seqfish_data_i = seqfish_data[(seqfish_data.cell == '5-0')]
axs[0].scatter(cell_mask_contour_i['x'], cell_mask_contour_i['y'], alpha=0.6, s=1, color='black')
axs[0].scatter(seqfish_data_i[(seqfish_data_i.gene == 'Aamp')]['x'], seqfish_data_i[(seqfish_data_i.gene == 'Aamp')]['y'], label='Original Points', alpha=0.6, s=5, color='red')
axs[0].scatter(cell_mask_contour_i['centerX'][0],cell_mask_contour_i['centerY'][0],color='green',s=100,marker='+')
axs[0].axis('equal')
axs[0].set_title('Original')
axs[1].scatter(seqfish_data_i[(seqfish_data_i.gene == 'Aamp')]['x_norm'], seqfish_data_i[(seqfish_data_i.gene == 'Aamp')]['y_norm'], label='Registered Points', alpha=0.6, s=5, color='red')
circle = plt.Circle((0, 0), 1, fill=False)
axs[1].add_artist(circle)
axs[1].set_title('Registered')
axs[1].scatter(0,0,color='green',s=100,marker='+')
axs[1].axis('equal')
axs[1].set_xlim(-1.5, 1.5)
axs[1].set_ylim(-1.5, 1.5)
plt.tight_layout()
plt.show()
Ignoring fixed x limits to fulfill fixed data aspect with adjustable data limits.
Ignoring fixed y limits to fulfill fixed data aspect with adjustable data limits.
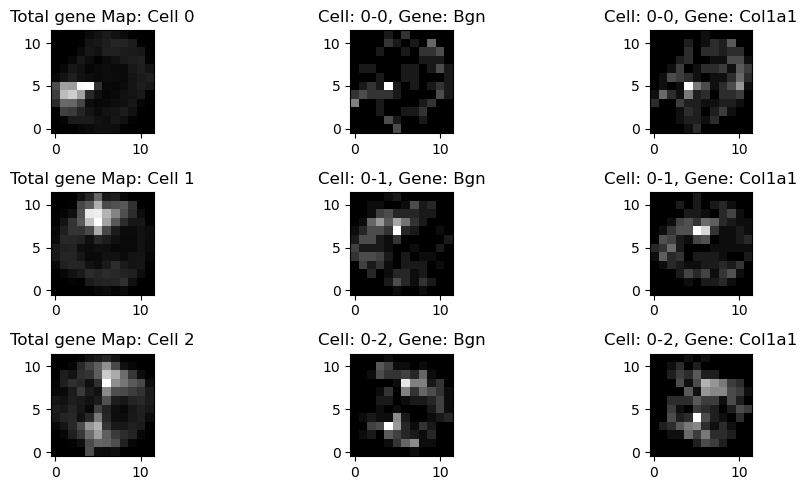
Represent the subcellular spatial expression of each cell and gene as a 48×48-pixel square image, and conduct resolution reduction
map_all_genes = get_gene_map(seqfish_data, cell_names, gene_names)
======> Loading transcripts file
1000 unique genes
171 unique cells
Converting to expression maps
100%|██████████| 171/171 [17:25<00:00, 6.12s/it]
print("map_all_genes shape:", map_all_genes.shape)
# np.savez_compressed(f"{data_path}{dataset}/cell_gene_map.npz", image = map_all_genes)
map_all_genes_low_res = map_all_genes.reshape(map_all_genes.shape[0], 12, 4, 12, 4,map_all_genes.shape[-1]).sum(axis=(2, 4))
print(map_all_genes_low_res.shape)
map_all_genes_low_res = np.transpose(map_all_genes_low_res, (0,3,1,2))
print("map_all_genes_low_res shape:", map_all_genes_low_res.shape)
# np.savez_compressed("f"{data_path}{dataset}/cell_gene_map_low_res.npz", image = map_all_genes_low_res)
map_all_genes shape: (171, 48, 48, 1000)
(171, 12, 12, 1000)
map_all_genes shape: (171, 1000, 12, 12)
fig, ax = plt.subplots(3, 3, figsize=(10, 5))
cell_id = [0,1,2]
i = 0
for cell in cell_id:
cell_gene = map_all_genes_low_res[cell]
cell_gene_sum = cell_gene.sum(axis = 0)
counts = np.sum(cell_gene, axis=(0, 1))
ax[i,0].imshow(cell_gene_sum, cmap='gray')
ax[i,0].set_title("Total gene Map: Cell " + str(cell))
ax[i,0].invert_yaxis()
ax[i,1].imshow(cell_gene[gene_names.index('Bgn'),:,:,], cmap='gray')
ax[i,1].set_title("Cell: " + cell_names[cell] + ", Gene: " + 'Bgn')
ax[i,1].invert_yaxis()
ax[i,2].imshow(cell_gene[gene_names.index('Col1a1'),:,:,], cmap='gray')
ax[i,2].set_title("Cell: " + cell_names[cell] + ", Gene: " + 'Col1a1')
ax[i,2].invert_yaxis()
i+=1
plt.tight_layout()
plt.show()
plt.close()