In silico predictions at subcellular resolution
import warnings
warnings.filterwarnings("ignore")
from model.train import *
import matplotlib.pyplot as plt
from sklearn.model_selection import KFold
data_path = './SVC/'
dataset = 'data/seqfish'
device = 'cuda:1'
gene_names = np.loadtxt(f'{data_path}{dataset}/gene_names.txt', dtype=str)
gene_names = gene_names.tolist()
cell_names = np.loadtxt(f'{data_path}{dataset}/cell_names.txt', dtype=str)
test_seqfish = np.load(f"{data_path}{dataset}/test_seqfish.npz") # 或 "cuda"
test_image = test_seqfish["data_ori"]
test_cell_morphology = test_seqfish["cell_morphology"]
test_nuclear_morphology = test_seqfish["nuclear_morphology"]
test_data_location = test_seqfish["location"]
test_cell_cycle_label = test_seqfish["identity_label"]
test_cell_cycle = test_seqfish["identity"]
test_cell_names = test_seqfish["cell_names"]
test_dataset = SVC_Dataset(
data_ori=test_image,
location=test_data_location,
cell_morphology_vec=test_cell_morphology,
nuclear_morphology_vec=test_nuclear_morphology,
identity_vec=test_cell_cycle_label,
)
print("number of testing cells:", len(test_dataset),', number of genes:', test_image.shape[1])
train_count_sum = np.load(f'{data_path}output/seqfish/train_count_sum.npy')
read_dir =f'{data_path}{dataset}/gene2vec_weight_seqfish.npy'
gene2vec_weight = torch.from_numpy(np.load(read_dir)).float()
val_loader = DataLoader(test_dataset, batch_size = 16, shuffle = False, num_workers = 4)
number of testing cells: 14 , number of genes: 1000
20-fold cross-prediction for validation
kf = KFold(n_splits=20, shuffle=True, random_state=2025)
groups = []
for train_index, test_index in kf.split(gene_names):
groups.append(np.array(gene_names)[test_index])
for i, group in enumerate(groups):
print(f"Group {i+1}: {group}")
Group 1: ['Abcf2' 'Actn1' 'Aldh9a1' 'Angptl2' 'Atp10a' 'Camsap2' 'Cherp' 'Csnk2a1'
'Ctnna1' 'Ddb1' 'Ehmt2' 'Eif2b5' 'Eif3d' 'Fbln5' 'Gtpbp1' 'Hipk2' 'Iars2'
'Ier5' 'Igf2bp2' 'Jdp2' 'Kpna6' 'Ldlr' 'Mlec' 'Myo1c' 'Napa' 'Ndel1'
'Nfkbia' 'Noc2l' 'Nsdhl' 'Nucks1' 'Pgd' 'Pgm1' 'Picalm' 'Pomgnt1'
'Ppp6r3' 'Rbpj' 'Rps6ka4' 'Smarca4' 'Socs5' 'Spry2' 'Ssrp1' 'Stk11'
'Stt3b' 'Sympk' 'Tcf20' 'Trp53' 'Tspan9' 'Uap1' 'Wwtr1' 'Zcchc14']
Group 2: ['Aff4' 'Anln' 'Arhgef17' 'Asns' 'Bak1' 'Bicc1' 'Bms1' 'Ccng1' 'Clstn1'
'Creb3l2' 'Cse1l' 'Cul1' 'Cyfip1' 'Cyp51' 'Dctn1' 'Elovl1' 'Fam32a'
'Fmr1' 'Gbf1' 'Hadh' 'Larp4' 'Lox' 'Lta4h' 'Mcm3' 'Mybbp1a' 'Myh10'
'Nasp' 'Ncapd2' 'Nfic' 'Nr2f2' 'Pdgfrb' 'Pigt' 'Polr2a' 'Rhoj' 'Rnh1'
'Ruvbl1' 'Sdc2' 'Sf3b3' 'Slit2' 'Smarcc2' 'Smc3' 'Steap2' 'Steap3'
'Tanc2' 'Tcf3' 'Tead1' 'Tomm40' 'Twf1' 'Uba1' 'Ube2g2']
Group 3: ['Aacs' 'Aldh1l2' 'Ankrd17' 'Cav1' 'Chaf1a' 'Champ1' 'Chpf' 'Col3a1'
'Col5a2' 'Ddx6' 'Ddx18' 'Dock7' 'Fam168b' 'Ftsj3' 'Galk1' 'Gas7' 'Gng12'
'Grn' 'Hdlbp' 'Hnrnph2' 'Hs2st1' 'Impdh1' 'Lbr' 'Mbnl1' 'Mprip' 'Ncaph2'
'Ndufs1' 'Nup54' 'P4hb' 'Pds5a' 'Pelp1' 'Prss23' 'Ptgis' 'Ptk2' 'Pycr2'
'Rbbp7' 'Rnf6' 'Rpl7l1' 'Rpn1' 'Sertad1' 'Sf3a2' 'Slc35e1' 'Smyd5'
'Tbrg4' 'Tfap4' 'Tmem109' 'Tspan5' 'Ube2z' 'Usp39' 'Wdr45b']
Group 4: ['Abhd17c' 'Actr1b' 'Anxa4' 'Cab39' 'Caprin1' 'Cdc42ep3' 'Cdk8' 'Coro1c'
'Ddost' 'E2f4' 'Efnb2' 'Etf1' 'Fam98a' 'Fgfr1' 'Ganab' 'Gna12' 'Hnrnpl'
'Igf2' 'Ilf3' 'Itgav' 'Lmf2' 'Ltbp1' 'Lzts2' 'Mapk1ip1l' 'Mllt1' 'Mpnd'
'Mthfd2' 'Mtmr2' 'Nid1' 'Palld' 'Rap2a' 'Rfc1' 'Rnf220' 'Samhd1' 'Shc1'
'Slc25a24' 'Slc38a4' 'Sp1' 'Srebf2' 'Srsf6' 'Ston1' 'Tgfbr3' 'Tnks1bp1'
'Ubr5' 'Utp20' 'Vcan' 'Wdr5' 'Wdr46' 'Wls' 'Zfp36l1']
Group 5: ['Atp13a3' 'Bcar1' 'Bclaf1' 'Bnc2' 'Cald1' 'Cd164' 'Chd1' 'Chtf8'
'Clptm1l' 'Col4a2' 'Cops8' 'Ddr2' 'Efhd2' 'Fam114a1' 'Fermt2' 'Fhl3'
'Fkbp5' 'Hipk1' 'Iglon5' 'Inppl1' 'Lurap1l' 'Macf1' 'Mical2' 'Mta1' 'Mvp'
'Nop14' 'Npc2' 'Nsun2' 'Plxnb2' 'Pold2' 'Poldip2' 'Ppp2r5d' 'Ptbp3'
'Pum2' 'Rpn2' 'Rybp' 'Scaf11' 'Setd3' 'Smg5' 'Snrnp70' 'Srrt' 'Ssr3'
'Stoml2' 'Supt5' 'Tmem259' 'Ube3c' 'Ubqln1' 'Wdr26' 'Wdr43' 'Xpot']
Group 6: ['Adamts5' 'Ap2a2' 'Atad2' 'Atp8b2' 'Bop1' 'Brd4' 'Carm1' 'Ccdc102a'
'Cnot1' 'Csnk1g2' 'Dag1' 'Dazap2' 'Dtl' 'Eftud2' 'Ezh2' 'Fam111a' 'Fstl1'
'Gfpt1' 'Gipc1' 'Gns' 'Heatr3' 'Map2k3' 'Mcl1' 'Mcm5' 'Mfsd5' 'Ncs1'
'Nfatc3' 'Nolc1' 'Obsl1' 'Osr1' 'Pef1' 'Phf23' 'Ppp1cc' 'Ppp1r15b'
'Ptpn9' 'Puf60' 'Rnf10' 'Sdc4' 'Sipa1l1' 'Snrnp200' 'Srpk1' 'Stat3'
'Tm4sf1' 'Tram1' 'Trio' 'Twsg1' 'Txnrd1' 'Ubqln4' 'Xbp1' 'Zfp91']
Group 7: ['Aaas' 'Abce1' 'Amfr' 'Arhgap11a' 'Arhgef1' 'Arid5b' 'Atf6b' 'Cdc42bpb'
'Cdh11' 'Chaf1b' 'Cops4' 'Dap' 'Drg2' 'Flii' 'Flnc' 'Furin' 'Glg1'
'Glyr1' 'Htra2' 'Insig1' 'Ipo8' 'Itpripl2' 'Leprot' 'Lrrc41' 'Mex3d'
'Msh2' 'Msrb3' 'Nfkb2' 'Nfya' 'Nisch' 'Nucb1' 'Numa1' 'Osbp' 'Pcbp1'
'Pcgf3' 'Pmpca' 'Ppp1r12a' 'Prmt3' 'Rab5b' 'Rad21' 'Ralb' 'Slc39a1'
'Smc4' 'Snx5' 'Surf4' 'Trpc4ap' 'Ubap2l' 'Ykt6' 'Zfhx3' 'Zmynd11']
Group 8: ['Aatf' 'Akap12' 'Anp32e' 'Ap2b1' 'Arhgap1' 'Axl' 'Bgn' 'Bin1' 'Brd3'
'Ccna2' 'Cdc42ep1' 'Cstf2t' 'Dlg1' 'Glipr2' 'Hmox1' 'Hnrnpd' 'Igfbp6'
'Il6st' 'Inf2' 'Ipo5' 'Kdm4a' 'Lonp1' 'Maged2' 'Myadm' 'Nab2' 'Ncbp1'
'Nudcd3' 'Nup107' 'Pck2' 'Pfkl' 'Plxna1' 'Ppp1r18' 'Psmd3' 'Samm50'
'Sdc1' 'Sfrp2' 'Slc7a1' 'Snx9' 'Sqstm1' 'Supt16' 'Tbl1x' 'Tjp2' 'Tk1'
'Tln1' 'Tnc' 'Tnfrsf1a' 'Tpr' 'Ube2i' 'Usp1' 'Xpnpep1']
Group 9: ['Abcf1' 'Acaca' 'Amotl1' 'Aplp2' 'Atf5' 'Bag6' 'Calu' 'Capn2' 'Cd109'
'Cdc16' 'Cdk16' 'Cdkn1a' 'Cdkn1b' 'Clic4' 'Dock1' 'Efr3a' 'Eif4g1' 'Elk3'
'Emilin2' 'Etv4' 'Fam3c' 'Fbln2' 'Fbxw8' 'Flna' 'Golt1b' 'Hspa4' 'Lbh'
'Lims1' 'Midn' 'Nap1l4' 'Ncor2' 'Pds5b' 'Pmp22' 'Pygb' 'Raf1' 'Rest'
'Sar1a' 'Scyl1' 'Sec31a' 'Setd7' 'Sipa1l3' 'Slc3a2' 'Soat1' 'Spr'
'Tmem214' 'Tpp2' 'Tulp4' 'Ubtf' 'Ung' 'Wwc2']
Group 10: ['Adamts1' 'Adipor1' 'Akt1' 'Aldh2' 'Anxa6' 'Ap1ar' 'Aqp1' 'Capza1'
'Casp3' 'Cbx5' 'Ccm2' 'Cdca8' 'Cited2' 'Dcakd' 'Eef1d' 'Eif6' 'Eps8'
'Fkbp10' 'Fn1' 'Foxm1' 'Iqgap1' 'Jmjd6' 'Lmnb1' 'Lrp1' 'Mapk6' 'Mat2a'
'Mtmr4' 'Myh9' 'Npnt' 'Pcolce' 'Pofut2' 'Ppp1ca' 'Prkar2a' 'Qrich1'
'Rgmb' 'Rpa2' 'Rrp12' 'S1pr3' 'Scarf2' 'Serpinh1' 'Setd5' 'Shoc2'
'Slco2a1' 'Trim35' 'Tsr1' 'Uba2' 'Uqcrc1' 'Utrn' 'Wdr6' 'Ythdf1']
Group 11: ['Aamp' 'Abl1' 'Acin1' 'Adm' 'Atic' 'Atp1b3' 'Cactin' 'Cic' 'Cluh'
'Csnk1d' 'Dctn2' 'Ddx27' 'Dnajc5' 'Dnmt1' 'Ehd1' 'Ext1' 'Fh1' 'Get4'
'Hdac7' 'Heatr5a' 'Igf1r' 'Lamc1' 'Lsm14a' 'Mark3' 'Mcm7' 'Metap1'
'Mki67' 'Nup205' 'Pacsin2' 'Pcnp' 'Plec' 'Prrc2a' 'Ptk7' 'Ptpn11' 'Qsox1'
'Rarg' 'Rpa1' 'Sav1' 'Sec14l1' 'Sec24d' 'Sf1' 'Sh3pxd2b' 'Smo' 'Tead2'
'Thoc5' 'Tm9sf3' 'Trim32' 'Vat1' 'Yap1' 'Zbtb7a']
Group 12: ['Api5' 'Arl2bp' 'Atxn2l' 'Ccdc6' 'Cmtm3' 'Col6a3' 'Dazap1' 'Dkc1'
'Eif2b1' 'Eml1' 'Gnai3' 'Gnl3l' 'Gtf2f1' 'Ing1' 'Ipo7' 'Kctd10' 'Lix1l'
'Lrrc59' 'Mcm6' 'Mpdz' 'Nfe2l1' 'Npepps' 'Nup153' 'Nxf1' 'Otud4' 'Ppan'
'Prkcsh' 'Prpf40a' 'Psme3' 'Psme4' 'Ptbp1' 'Pus1' 'Rab5c' 'Rab10' 'Rrn3'
'Rtcb' 'Sephs2' 'Serpinf1' 'Slc2a1' 'Slc7a5' 'Snx17' 'Sp3' 'Strap'
'Suclg2' 'Supt6' 'Tgfb1i1' 'Tmed7' 'Uso1' 'Xpo5' 'Zfp36l2']
Group 13: ['Actr1a' 'Aebp1' 'Ano6' 'Arhgef40' 'Birc6' 'Cad' 'Ccnd1' 'Cdh2' 'Col1a1'
'Colec12' 'Dusp7' 'Dysf' 'Eif2s1' 'Filip1l' 'Fxr1' 'Gas1' 'Gnl2' 'Hnrnpf'
'Hyou1' 'Ide' 'Kif1c' 'Larp4b' 'Mgat2' 'Mmp14' 'Mogs' 'Nfat5' 'Nfe2l2'
'Nmt2' 'Nop2' 'Nploc4' 'Nr2f1' 'Nup85' 'Papss1' 'Pkd2' 'Pkn1' 'Pola2'
'Prrx1' 'Ranbp3' 'Sh3pxd2a' 'Sipa1l2' 'Slc48a1' 'Smarcc1' 'Snw1' 'Sox9'
'Tmem97' 'Tnks' 'Topbp1' 'Usp5' 'Usp7' 'Wnk1']
Group 14: ['Ago2' 'Anapc4' 'Arf6' 'Ccne1' 'Cdca4' 'Cep170' 'Ckap4' 'Csrp1' 'Ctsd'
'Cul4a' 'Dse' 'Eif3l' 'Elp2' 'Emp1' 'Fam171a1' 'Idh2' 'Kctd17' 'Klhdc3'
'Lasp1' 'Ltbp2' 'M6pr' 'Map4k4' 'Maz' 'Mcm2' 'Mcm4' 'Megf8' 'Ncln'
'Nr2f6' 'Pdia6' 'Ppp2ca' 'Prep' 'R3hdm4' 'Rras2' 'Rrm1' 'Ryk' 'Sfxn3'
'Slc38a2' 'St3gal2' 'Stt3a' 'Tax1bp1' 'Tbl3' 'Tnpo3' 'Tnrc18' 'Top2a'
'Trim8' 'Tulp3' 'Usp9x' 'Vasn' 'Vcl' 'Zcchc24']
Group 15: ['Alkbh5' 'Ankrd13a' 'Arpc4' 'Cdc37' 'Cnpy3' 'Col5a1' 'Col16a1' 'Copa'
'Cotl1' 'Cttn' 'Ddx23' 'Degs1' 'Dpp9' 'Dpy19l1' 'Dynll2' 'Egln2'
'Ehbp1l1' 'Emb' 'Fads1' 'Fasn' 'Fbn1' 'Fmnl2' 'Fosl2' 'Gart' 'Gtf2i'
'Immt' 'Lpp' 'Lrpprc' 'Mapkapk2' 'Naa50' 'Otub1' 'Pcdh19' 'Pitrm1' 'Pnn'
'Ppfibp1' 'Rraga' 'Sec61a1' 'Serp1' 'Slc16a1' 'Smarcb1' 'Snd1' 'Spop'
'Thbd' 'Trip12' 'Tspyl1' 'Txlna' 'Usp4' 'Vasp' 'Wdfy3' 'Zfp598']
Group 16: ['Acly' 'Arhgap10' 'Atl3' 'Atn1' 'Bptf' 'Cap1' 'Cdc42ep2' 'Clip2' 'Col6a2'
'Copb2' 'Copz1' 'Crim1' 'Ddah1' 'Ddx21' 'Dnaja3' 'Dync1li2' 'Eif3b'
'Ergic1' 'Flnb' 'Foxk2' 'Grb10' 'Hif1a' 'Ipo13' 'Itgb1' 'Klhl9' 'Kpna4'
'Kpnb1' 'Mast2' 'Med13l' 'Mtap' 'Naa25' 'Pip4k2b' 'Polr2b' 'Ptpn21'
'Pttg1ip' 'Pxdn' 'Rad23b' 'Ranbp2' 'Ruvbl2' 'Senp3' 'Serpine1' 'Shmt1'
'Smchd1' 'Smg7' 'Srsf1' 'Stk16' 'Tagln' 'Trrap' 'Xrn2' 'Zmiz1']
Group 17: ['Brd2' 'Cd248' 'Copb1' 'Dlk1' 'Dnttip2' 'Drosha' 'Ebna1bp2' 'Fam120a'
'Fkbp8' 'Gps1' 'Hdac3' 'Ints5' 'Ist1' 'Lpar1' 'Man1a2' 'Mrpl37' 'Mta2'
'Nadk' 'Nol11' 'Paip2b' 'Phlda3' 'Plbd2' 'Postn' 'Prc1' 'Prkci' 'Prmt7'
'Psat1' 'Psmd8' 'Pum1' 'Pwp1' 'Pygo2' 'Rbfox2' 'Rbm10' 'Rcn1' 'Rhob'
'Sec23a' 'Sgk1' 'Slc25a39' 'Smc1a' 'Snai1' 'Srp68' 'Tbrg1' 'Tcof1'
'Tfdp1' 'Tns3' 'U2af2' 'Ugdh' 'Usp47' 'Wdr1' 'Zfp462']
Group 18: ['Actl6a' 'Akap8' 'Aldh18a1' 'Amotl2' 'Ap3d1' 'Arhgdia' 'Asap1' 'Axin1'
'Cand1' 'Ccnk' 'Cdc5l' 'Cdk1' 'Ctdsp1' 'Cyb5b' 'Etv5' 'G6pdx' 'Galnt1'
'Golim4' 'Gorasp2' 'Grk6' 'Hnrnpm' 'Incenp' 'Klhl5' 'Loxl3' 'Maea'
'Map3k3' 'Morc2a' 'Myo1b' 'Myof' 'Nacc1' 'Pam' 'Pcdh18' 'Pdia4' 'Ppm1g'
'Pprc1' 'Prkaca' 'Ptx3' 'Racgap1' 'Rangap1' 'Rassf8' 'Raver1' 'Rbbp4'
'Rhoq' 'Rragc' 'Scaf8' 'Shmt2' 'Slc38a10' 'Tcf19' 'Tnk2' 'Yif1b']
Group 19: ['Actn4' 'Adam9' 'Add1' 'Ahcyl1' 'Ap1m1' 'Arcn1' 'Atp1a1' 'Bub3' 'Col6a1'
'Coro1b' 'Cs' 'Cyb5r3' 'Ddx3x' 'Ddx46' 'Dhx15' 'Dok1' 'Eif4h' 'Farp1'
'Fhdc1' 'Fkbp9' 'Fxr2' 'Glud1' 'Hmgcr' 'Isg20l2' 'Kat2a' 'Kcmf1' 'Lrrc42'
'Nek7' 'Nmnat2' 'Nt5dc2' 'Nup155' 'Pcyox1' 'Pdia3' 'Pdxdc1' 'Pgrmc2'
'Phldb2' 'Prcc' 'Rab31' 'Safb' 'Ski' 'Smarce1' 'Smc6' 'Srrm1' 'Syde1'
'Tab2' 'Timp3' 'Trim28' 'Vgll3' 'Zdhhc5' 'Zyx']
Group 20: ['Acat1' 'Agpat1' 'Anapc1' 'Anapc2' 'Bckdk' 'Cd44' 'Cdk14' 'Clint1'
'Cyth3' 'Dbnl' 'Dcaf7' 'Dhcr24' 'Dhx9' 'Dlst' 'Eif4ebp2' 'Fat1' 'Fscn1'
'Gsn' 'Hectd1' 'Hspg2' 'Irs1' 'Khsrp' 'Larp1' 'Lhfpl2' 'Lrp6' 'Man2a1'
'Mbtps1' 'Med25' 'Msn' 'Myc' 'Nup188' 'Pak2' 'Papola' 'Poldip3' 'Prpf8'
'Rhobtb3' 'Rrs1' 'Scaf1' 'Slc25a1' 'Slk' 'Sqle' 'Stip1' 'Synpo' 'Tagln2'
'Thbs1' 'Thrap3' 'Tpbg' 'Tsc22d2' 'Xpo1' 'Zfp106']
ckpt_dir = f"{data_path}/checkpoints/"
ckpt = torch.load(ckpt_dir +'SVC_seqfish.pth', map_location=device)
state = ckpt.get("model_state_dict", ckpt)
state = {k.removeprefix("module."): v for k, v in state.items()}
model = SVC(
gene2vec_weight = gene2vec_weight,
cell_identity_dim = test_cell_cycle_label.shape[1],
).to(device)
model.load_state_dict(state)
<All keys matched successfully>
save the mean and dispersion paramater for each pixel, and set the background pixel values to 0
prediction_impute_mu = torch.zeros(test_image.shape).to(device)
prediction_impute_r = torch.zeros(test_image.shape).to(device)
background_pixel = torch.Tensor([[0,0],[0,1],[0,10],[0,11],[1,0],[1,11],[10,0],[10,11],[11,0],[11,1],[11,10],[11,11]]).to(device).to(torch.long)
model.eval()
for selected_gene in groups:
gene_to_impute = [gene_names.index(i) for i in selected_gene if i in gene_names]
gene_not_to_impute = [gene for gene in range(len(gene_names)) if gene not in gene_to_impute]
predictions_mu= []
predictions_r = []
with torch.no_grad():
for i, (inputs_ori, cell_morphology, nuclear_morphology, location, cell_cycle) in enumerate(val_loader, 0):
inputs_ori = inputs_ori.to(device).float()
location = location.to(device).float()
cell_morphology = cell_morphology.to(device).float()
nuclear_morphology = nuclear_morphology.to(device).float()
cell_cycle = cell_cycle.to(device).float()
inputs_ori_mask = inputs_ori.clone()
inputs_ori_mask[:,gene_to_impute] = 0
cell_median_train = np.median((train_count_sum[:,gene_not_to_impute]).sum(axis=1))
size_factor = (inputs_ori_mask.sum((1,2,3))/cell_median_train).unsqueeze(1).unsqueeze(2).unsqueeze(3)
inputs = inputs_ori / size_factor # (n_cell, n_gene, 12, 12)
mask = torch.zeros(inputs.shape[0], inputs.shape[1]).to(device).bool()
mask[:,gene_to_impute] = True
inputs_mask = inputs.clone()
inputs_mask[:,gene_to_impute] = 0
_, predicts_mu, predicts_r= model(inputs_mask, mask, location, cell_morphology, nuclear_morphology, cell_cycle)
predicts_mu = predicts_mu * size_factor # batch*gene*h*w
predictions_mu.append(predicts_mu)
predictions_r.append(predicts_r)
predictions_mu = torch.cat(predictions_mu, dim=0)
predictions_r = torch.cat(predictions_r, dim=0)
predictions_mu[:,:,background_pixel[:,0],background_pixel[:,1]] = 0
predictions_r[:,:,background_pixel[:,0],background_pixel[:,1]] = 0
prediction_impute_mu[:,gene_to_impute] = predictions_mu[:,gene_to_impute]
prediction_impute_r[:,gene_to_impute] = predictions_r[:,gene_to_impute]
# np.savez_compressed(f'{data_path}output/seqfish/prediction_all_genes_mu.npz', prediction=prediction_impute_mu.cpu().numpy())
# np.savez_compressed(f'{data_path}output/seqfish/prediction_all_genes_r.npz', prediction=prediction_impute_r.cpu().numpy())
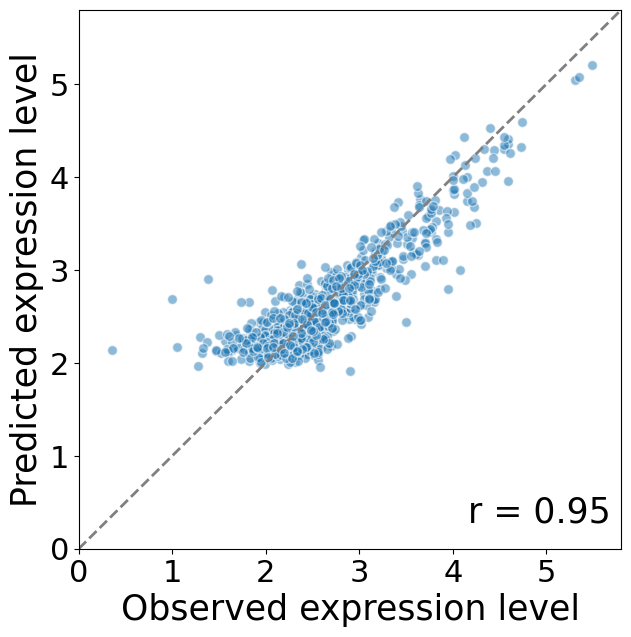
first compare the predicted cellular expression values vs ground truth at single-cell level
pred_exp_level_SVC = (prediction_impute_mu.cpu().numpy().sum((-1,-2))).mean(0)
exp_level = (test_image.sum((-1,-2))).mean(0)
fig, ax = plt.subplots(1, 1, figsize=(7, 7))
plt.scatter(np.log1p(exp_level), np.log1p(pred_exp_level_SVC), s=50, alpha=0.5,edgecolors='white')
plt.plot([0, 6], [0, 6], '--', lw=2, color='gray')
plt.xlim(0, 5.8)
plt.ylim(0, 5.8)
ax.set_xticks([0,1,2,3,4,5])
ax.set_xticklabels([0,1,2,3,4,5],fontsize=22)
ax.set_yticks([0,1,2,3,4,5])
ax.set_yticklabels([0,1,2,3,4,5],fontsize=22)
plt.xlabel('Observed expression level',fontsize=25)
plt.ylabel('Predicted expression level',fontsize=25)
ax.text(0.98, 0.05, f'r = {np.corrcoef(exp_level, pred_exp_level_SVC)[0, 1]:.2f}', transform=ax.transAxes, ha='right',fontsize=25)
plt.show()
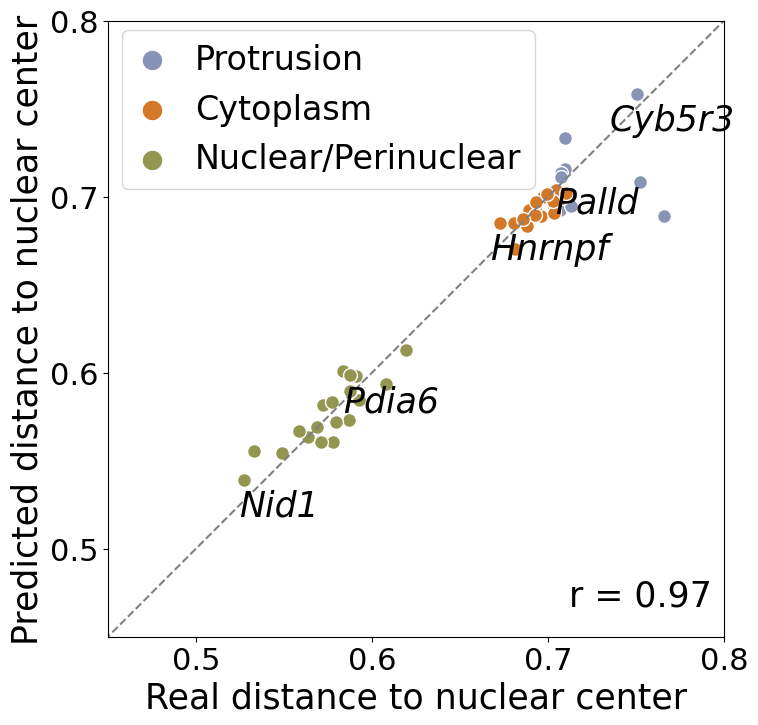
focus on the 49 well-annotated genes
# seqfish+ genes in extended data fig 3
protrusionList = ['Cyb5r3', 'Sh3pxd2a', 'Ddr2', 'Kif1c', 'Kctd10',
'Dynll2', 'Arhgap11a', 'Dync1li2', 'Palld', 'Naa50']
nuclearList = ['Col1a1', 'Fn1', 'Fbln2', 'Col6a2', 'Bgn',
'Nid1', 'Lox', 'P4hb', 'Aebp1', 'Emp1',
'Col5a1', 'Sdc4', 'Postn', 'Col3a1', 'Pdia6',
'Col5a2', 'Itgb1', 'Calu', 'Pdia3']
cytoplasmList = ['Ddb1', 'Myh9', 'Actn1', 'Tagln2', 'Kpnb1',
'Hnrnpf', 'Ppp1ca', 'Hnrnpl', 'Pcbp1', 'Tagln',
'Fscn1', 'Psat1', 'Cald1', 'Snd1', 'Uba1',
'Hnrnpm', 'Cap1', 'Ssrp1', 'Ugdh', 'Caprin1']
selected_gene = protrusionList + nuclearList + cytoplasmList
selected_gene_idx = [gene_names.index(i) for i in selected_gene]
print("gene to impute:",selected_gene_idx)
gene to impute: [203, 775, 219, 421, 417, 250, 63, 249, 586, 522, 168, 319, 299, 174, 92, 546, 443, 582, 24, 279, 171, 753, 635, 169, 601, 172, 411, 108, 599, 217, 516, 14, 865, 427, 374, 639, 376, 590, 864, 323, 662, 107, 817, 934, 377, 111, 842, 945, 113]
calculate pred and observed relative distance to nuclear center for each gene
prediction_gene_dist = np.zeros((len(test_dataset),len(selected_gene_idx)))
truth_gene_dist = np.zeros((len(test_dataset),len(selected_gene_idx)))
I = 0
for cell in range(len(test_dataset)):
cell_gene_pred = prediction_impute_mu[cell].cpu().numpy()
cell_gene_real = test_image[cell]
J = 0
for gene in selected_gene_idx:
heatmap_pred = cell_gene_pred[gene,:,:,]
heatmap_real = cell_gene_real[gene,:,:,]
prediction_gene_dist[I,J] = compute_relative_dist_to_nuclear_center(heatmap_pred)
truth_gene_dist[I,J] = compute_relative_dist_to_nuclear_center(heatmap_real)
J += 1
I += 1
gene_to_impute_type = []
for i in selected_gene:
if i in protrusionList:
gene_to_impute_type.append('Protrusion')
elif i in cytoplasmList:
gene_to_impute_type.append('Cytoplasm')
else:
gene_to_impute_type.append('Nuclear/Perinuclear')
gene_to_impute_type_dict = {'Protrusion':1,'Cytoplasm':2,'Nuclear/Perinuclear':3}
gene_to_impute_type_num = np.array([gene_to_impute_type_dict[i] for i in gene_to_impute_type])
prediction_gene_dist_mean = np.nanmean(prediction_gene_dist,axis=0)
prediction_gene_dist_mean = np.array(prediction_gene_dist_mean).reshape(-1,1)
truth_gene_dist_mean = np.nanmean(truth_gene_dist,axis=0)
truth_gene_dist_mean = np.array(truth_gene_dist_mean).reshape(-1,1)
impute_real_concat = np.concatenate((truth_gene_dist_mean,prediction_gene_dist_mean), axis=1)
representative_genes = ['Nid1', 'Pdia6', "Hnrnpf", "Palld", "Cyb5r3"]
fig, ax = plt.subplots(1, 1, figsize=(8, 8))
colors = prediction_gene_dist_mean
custom_colors = ["#8794b6","#d47828","#939650"]
i=0
for type in gene_to_impute_type_dict:
ax.scatter(impute_real_concat[np.array(gene_to_impute_type) == type,0], impute_real_concat[np.array(gene_to_impute_type) == type,1],c=custom_colors[i],s=100,label=type,edgecolor='white')
i+=1
ax.legend(title='',fontsize=24,bbox_to_anchor=(0.72, 0.7),markerscale=1.5,handlelength=1)
ax.text(0.98, 0.05, f'r = {np.corrcoef(impute_real_concat[:,0], impute_real_concat[:,1])[0, 1]:.2f}', transform=ax.transAxes, ha='right',fontsize=25)
for gene in selected_gene:
if gene in representative_genes:
ax.annotate(gene, (impute_real_concat[selected_gene.index(gene),0]+0.02, impute_real_concat[selected_gene.index(gene),1]-0.001), textcoords="offset points", xytext=(0,-25), ha='center', fontsize=25,fontstyle="italic")
ax.set_aspect('equal')
ax.plot([0, 0.8], [0, 0.8], '--', color='gray')
ax.set_xlim(0.45, 0.8)
ax.set_ylim(0.45, 0.8)
ax.set_xticks([0.5,0.6,0.7,0.8])
ax.set_xticklabels([0.5,0.6,0.7,0.8],fontsize=22)
ax.set_yticks([0.5,0.6,0.7,0.8])
ax.set_yticklabels([0.5,0.6,0.7,0.8],fontsize=22)
ax.set_xlabel('Real distance to nuclear center', fontsize=25)
ax.set_ylabel('Predicted distance to nuclear center', fontsize=25)
plt.show()
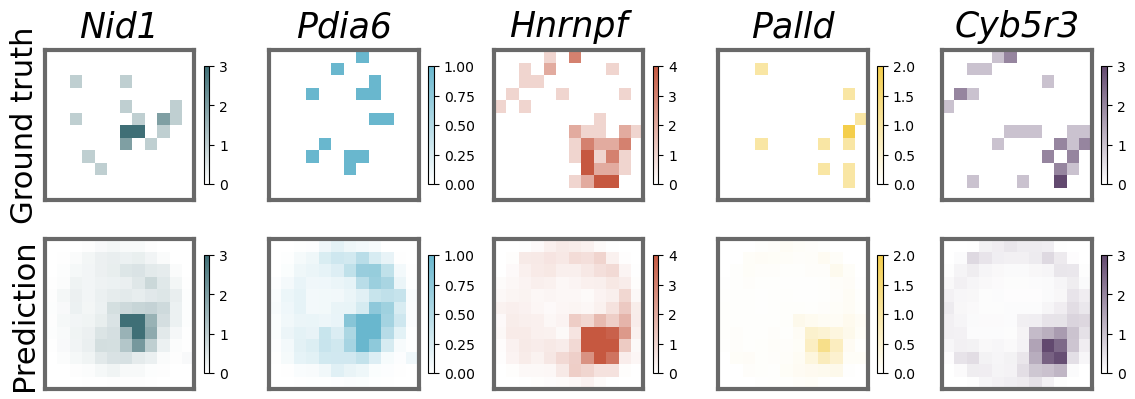
pred and observed subcellular pattern for representative genes
colors = ['#3f6f76', '#69b7ce', '#c65840', '#f4ce4b', '#62496f']
custom_cmap = [create_white_to_color_cmap(i) for i in colors]
gene_set = ["Nid1", "Pdia6", "Hnrnpf", "Palld", "Cyb5r3"]
cell = '16-3'
cell_idx = list(test_cell_names).index(cell)
fig, ax = plt.subplots(2, 5, figsize=(14, 4.5), gridspec_kw={ "hspace": 0.2})
i = 0
for gene in gene_set:
cell_gene = prediction_impute_mu[cell_idx].cpu().numpy()
cell_gene_real = test_image[cell_idx]
vmax = cell_gene_real[gene_names.index(gene),:,:,].max()
cell_gene_sum = cell_gene.sum(axis = 0)
ax0 = ax[0,i].imshow(cell_gene_real[gene_names.index(gene),:,:,], cmap=custom_cmap[i],vmin=0,vmax=vmax)#,vmin=-0.2,vmax=15)
if i == 0:
ax[i,0].set_ylabel("Ground truth",fontsize=22)
ax[0,i].set_title(f"{gene_names[gene_names.index(gene)]}",fontsize=25,pad=10,fontstyle='italic')
cbar = plt.colorbar(ax0,shrink=0.75)
ax[0,i].invert_yaxis()
for spine in ax[0,i].spines.values():
spine.set_linewidth(3)
spine.set_color('dimgrey')
ax1 = ax[1,i].imshow(cell_gene[gene_names.index(gene),:,:,], cmap=custom_cmap[i],vmin=0,vmax=vmax)
cbar = plt.colorbar(ax1,shrink=0.75)
if i == 0:
ax[1,i].set_ylabel(f"Prediction",fontsize=22)
ax[1,i].invert_yaxis()
for spine in ax[1,i].spines.values():
spine.set_linewidth(3)
spine.set_color('dimgrey')
ax[0,i].set_xticks([]) # 隐藏 x 轴刻度
ax[0,i].set_yticks([])
ax[1,i].set_xticks([])
ax[1,i].set_yticks([])
i+=1
plt.tight_layout()
plt.show()
plt.close()
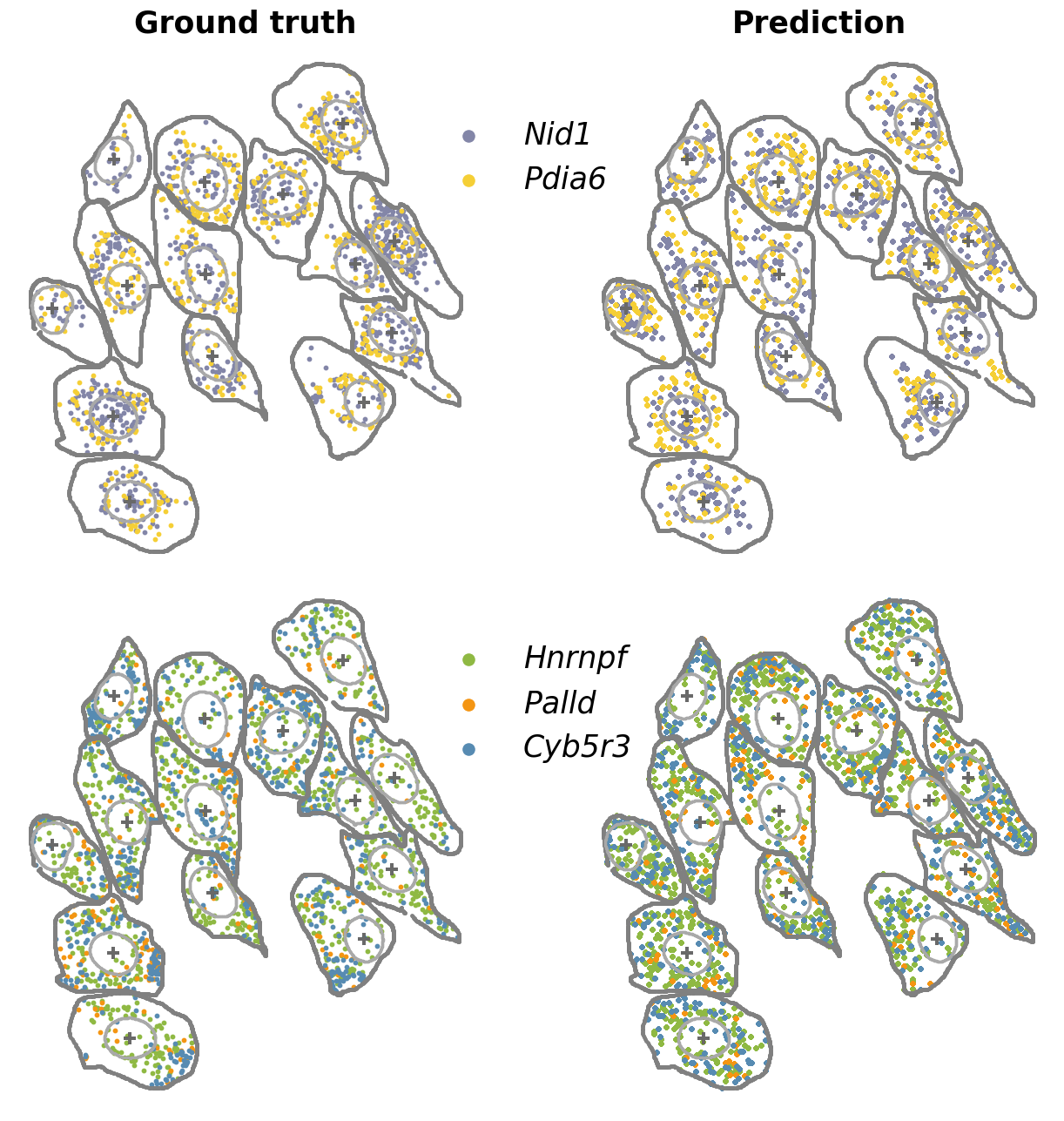
postprocessing: map the predicted high-resolution spatial expression from the unified cellular coordinate system back to the original cell morphology
NB sampling
calculating the relative radial distance and the angular position for each pixel
identifying the cellular boundary point corresponding to the same angle
determining the Cartesian coordinates of the transcript within the original cell
prediction_49_genes_mu = prediction_impute_mu[:,selected_gene_idx].cpu().numpy()
prediction_49_genes_r = prediction_impute_r[:,selected_gene_idx].cpu().numpy()
# np.savez_compressed(f'{data_path}output/seqfish/prediction_49_genes_mu.npz', prediction=prediction_49_genes_mu)
# np.savez_compressed(f'{data_path}output/seqfish/prediction_49_genes_r.npz', prediction=prediction_49_genes_r))
count_data = postprocess_sampling(prediction_49_genes_mu, prediction_49_genes_r, seed=2025)
print("shape of count_data:",count_data.shape)
100%|██████████| 14/14 [00:00<00:00, 123.91it/s]
shape of count_data: (14, 49, 48, 48)
predictions_pixel = postprocess_predictions(count_data, selected_gene, test_cell_names)
df_cell_contour = pd.read_pickle(f"{data_path}{dataset}/cell_mask_contour_preprocessed.pkl")
print(df_cell_contour.head())
df_nuclear_contour = pd.read_pickle(f"{data_path}{dataset}/nuclear_mask_contour_preprocessed.pkl")
print(df_nuclear_contour.head())
cell x y centerX centerY direction_vec distance_to_center
0 0-0 521 496 1079 724 -158.0 602.783543
1 0-0 519 496 1079 724 -158.0 604.635427
2 0-0 517 494 1079 724 -157.5 607.242950
3 0-0 515 494 1079 724 -158.0 609.094410
4 0-0 513 492 1079 724 -157.5 611.702542
cell x y centerX centerY
0 0-0 1051 631 1079 724
1 0-0 1050 632 1079 724
2 0-0 1049 632 1079 724
3 0-0 1048 632 1079 724
4 0-0 1047 632 1079 724
predictions_pixel = postprocess_predictions_original(predictions_pixel, df_cell_contour)
print(predictions_pixel.head())
# predictions_pixel.to_csv(f"{data_path}output/seqfish/prediction_count_data_49_genes.csv")
100%|██████████| 14/14 [00:05<00:00, 2.58it/s]
cell gene count x y ratio direction_vec centerX centerY \
0 1-3 Cyb5r3 1.0 25 0 0.981159 -86.5 1026 1173
1 1-3 Cyb5r3 1.0 27 1 0.948775 -81.0 1026 1173
2 1-3 Cyb5r3 1.0 34 1 1.000000 -65.0 1026 1173
3 1-3 Cyb5r3 1.0 35 4 0.943269 -59.5 1026 1173
4 1-3 Cyb5r3 1.0 22 5 0.773363 -94.5 1026 1173
distance_to_center angle_radians x_original y_original
0 161.208985 -1.509710 1035.841573 1012.091702
1 163.464839 -1.413717 1051.571535 1011.547685
2 287.200279 -1.134464 1147.376082 912.708151
3 230.041701 -1.038471 1142.754988 974.789363
4 127.292819 -1.649336 1016.012721 1046.099582
visualization at original scale
data = pd.read_pickle(f"{data_path}{dataset}/seqfish_data_dict.pkl")
seqfish_data = data['data_df']
print(seqfish_data.head())
x y gene cell nucleus batch umi centerX \
0 1217.437557 557.583252 4933401b06rik 5-0 -1 0 1 1003
1 1096.190309 394.835294 4933401b06rik 5-0 5 0 1 1003
2 1093.189494 572.832405 4933401b06rik 5-0 -1 0 1 1003
3 1005.120220 297.196271 4933401b06rik 5-0 -1 0 1 1003
4 1142.815026 378.376491 4933401b06rik 5-0 -1 0 1 1003
centerY type sc_total
0 425 fibroblast 32224
1 425 fibroblast 32224
2 425 fibroblast 32224
3 425 fibroblast 32224
4 425 fibroblast 32224
preserve_idx_cell_mask_contour =[]
for i in df_cell_contour.cell:
if i in test_cell_names:
preserve_idx_cell_mask_contour.append(True)
else:
preserve_idx_cell_mask_contour.append(False)
preserve_idx_nuclear_mask_contour =[]
for i in df_nuclear_contour.cell:
if i in test_cell_names:
preserve_idx_nuclear_mask_contour.append(True)
else:
preserve_idx_nuclear_mask_contour.append(False)
preserve_idx_seqfish_data =[]
for i in seqfish_data.cell:
if i in test_cell_names:
preserve_idx_seqfish_data.append(True)
else:
preserve_idx_seqfish_data.append(False)
def plot_boundary(ax, df_cell_contour_i, df_nuclear_contour_i):
ax.scatter(df_cell_contour_i.x, df_cell_contour_i.y, color="grey", s=6, zorder=20)
ax.scatter(df_nuclear_contour_i.x, df_nuclear_contour_i.y, alpha=0.6, s=2, color="darkgrey", zorder=20)
ax.scatter(df_nuclear_contour_i["centerX"], df_nuclear_contour_i["centerY"],
marker="+", color="dimgray", s=100, zorder=20)
ax.set_aspect("equal")
ax.axis("off")
def plot_gene_points(ax, df, genes, colors, xcol, ycol, label_style="italic", legend=True):
for g, c in zip(genes, colors):
sub = df[df["gene"] == g]
ax.scatter(sub[xcol], sub[ycol], s=10, color=c, label=g)
leg = None
if legend:
leg = ax.legend(fontsize=25, markerscale=3, frameon=False)
if label_style == "italic" and leg is not None:
for t in leg.get_texts():
t.set_fontstyle("italic")
return leg
def plot_truth_pred_pair(ax_truth, ax_pred,
truth_df, pred_df_exploded,
genes, colors,
df_cell_contour_i, df_nuclear_contour_i,
title_truth=None, title_pred=None,
legend_anchor_truth=None, legend_anchor_pred=None):
# truth
plot_boundary(ax_truth, df_cell_contour_i, df_nuclear_contour_i)
plot_gene_points(ax_truth, truth_df, genes, colors, xcol="x", ycol="y")
if title_truth:
ax_truth.set_title(title_truth, fontsize=25, fontweight="bold")
if legend_anchor_truth is not None:
ax_truth.legend_.set_bbox_to_anchor(legend_anchor_truth)
# pred
plot_boundary(ax_pred, df_cell_contour_i, df_nuclear_contour_i)
plot_gene_points(ax_pred, pred_df_exploded, genes, colors, xcol="x_original", ycol="y_original", legend=False)
if title_pred:
ax_pred.set_title(title_pred, fontsize=25, fontweight="bold")
if legend_anchor_pred is not None:
ax_pred.legend_.set_bbox_to_anchor(legend_anchor_pred)
truth_i = seqfish_data[preserve_idx_seqfish_data]
df_cell_contour_i = df_cell_contour[preserve_idx_cell_mask_contour]
df_nuclear_contour_i = df_nuclear_contour[preserve_idx_nuclear_mask_contour]
pred_exploded = predictions_pixel.loc[predictions_pixel.index.repeat(predictions_pixel['count'])].reset_index(drop=True)
# ---- plotting ----
fig, ax = plt.subplots(2, 2, figsize=(17, 16), gridspec_kw={"hspace": 0., "wspace": 0.})
genes1 = ["Nid1", "Pdia6"]
plot_truth_pred_pair(
ax[0, 0], ax[0, 1],
truth_df=truth_i, pred_df_exploded=pred_exploded,
genes=genes1, colors=["#8386a8", "#f5cf36"],
df_cell_contour_i=df_cell_contour_i, df_nuclear_contour_i=df_nuclear_contour_i,
title_truth="Ground truth", title_pred="Prediction",
legend_anchor_truth=(0.85, 0.9)
)
genes2 = ["Hnrnpf", "Palld", "Cyb5r3"]
plot_truth_pred_pair(
ax[1, 0], ax[1, 1],
truth_df=truth_i, pred_df_exploded=pred_exploded,
genes=genes2, colors=["#8fb943", "#f49512", "#578bb2"],
df_cell_contour_i=df_cell_contour_i, df_nuclear_contour_i=df_nuclear_contour_i,
legend_anchor_truth=(0.85, 0.6)
)
plt.show()